








 253 / 2894
253 / 2894

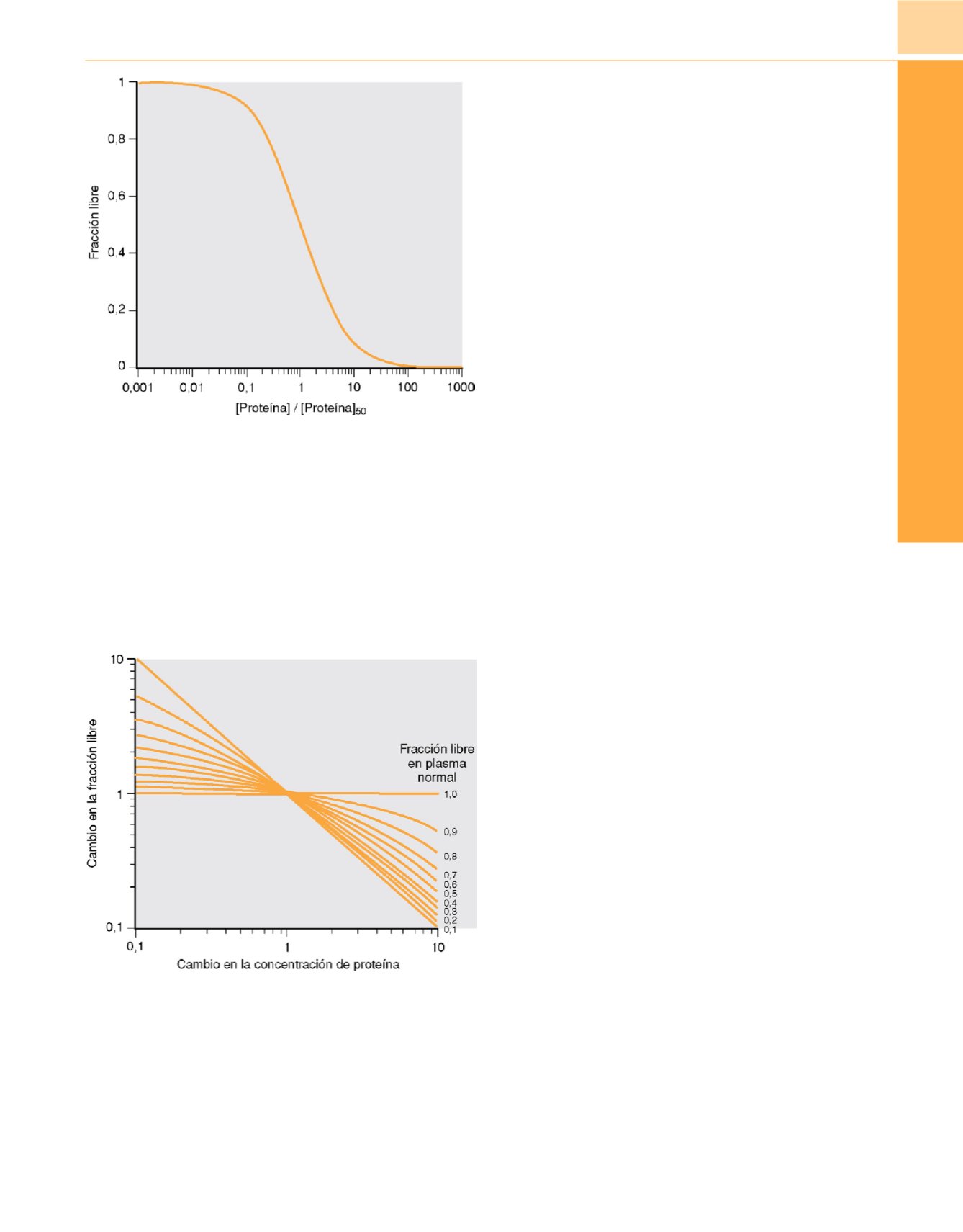
La
figura 9-11muestra la relación entre cambios en la concentra-
ción de proteína y cambios en la fracción de fármaco libre para
fármacos de uso clínico habitual con diferentes grados de unión a
proteínas. Las líneas de la gráfica corresponden a diferentes fárma-
cos cuya fracción libre en plasma normal oscila entre 1 (línea
horizontal) y aproximadamente 0 (línea diagonal recta).
Para fármacos no unidos (fracción libre=1), no existe rela-
ción entre fracción libre y concentración de proteína, como indica
la línea horizontal plana. Para fármacos cuya fracción libre es habi-
tualmente del 90%, se produce un cambio leve en la concentración
de fármaco libre con los cambios en la concentración de proteína.
Para fármacos que se unen en gran proporción a proteínas, la
concentración de fármaco libre cambia de modo casi inverso al
cambio en la concentración de proteína. Conforme el porcentaje
de unión se aproxima al 100% (fármaco libre
→
0) la relación entre
cambio en la concentración de proteína y cambio en la fracción
libre se vuelve inversamente proporcional. Recuerde que nunca hay
un cambio mayor que el proporcional en la concentración de
fármaco libre al cambiar la concentración plasmática. Por ejemplo,
lo más que produce un cambio del 10% en la concentración de
proteína es un cambio del 10% en la concentración de fármaco
libre, y sólo en el caso de que el fármaco estuviera unido casi al
100% a proteínas plasmáticas.
La observación in vitro de que el cambio en la concentración
de proteína modifica la concentración de fármaco libre, como
indica la
figura 9-11 ,no se aplica necesariamente in vivo. Es el
fármaco libre (es decir, no unido a proteínas) el que se equilibra
entre el plasma y los tejidos. Si disminuye la unión a proteínas
plasmáticas, el gradiente de concentración de fármaco libre entre
el plasma y los tejidos periféricos aumenta. Como consecuencia,
cuando la unión a proteínas disminuye, se alcanza un equilibrio
entre la concentración de fármaco libre en plasma y tejidos con
una concentración plasmática total de fármaco inferior. Esta menor
concentración produce la apariencia de que el fármaco se ha dis-
tribuido en un espacio total más amplio. El descenso de la unión a
proteínas produce un aumento del volumen de distribución apa-
rente cuando se relaciona con la concentración total de fármaco,
en lugar de con la concentración de fármaco libre.
El aumento del volumen de distribución en equilibrio, Vd
ee
,
observado con una menor unión a proteínas plasmáticas es básica-
mente una ilusión. Si sólo midiéramos la concentración de fármaco
no unido, no habría apenas cambio en el volumen de distribución
aparente de fármacos lipófilos (como la mayoría de los utilizados
en anestesia) porque es el reparto del fármaco en los tejidos peri-
féricos el que determina principalmente la concentración libre de
los fármacos lipófilos.
Los cambios en la unión a proteínas pueden influir en la
depuración de los fármacos. Si un fármaco tiene un índice de extrac-
ción alto, el hígado extraerá casi todo el fármaco que le llega, con
independencia del grado de unión a proteínas. Sin embargo, si el
fármaco tiene un índice de extracción hepático bajo, un aumento de
la fracción de fármaco libre aumenta el gradiente propulsor con un
incremento asociado de la depuración. La unión a proteínas afecta
también a la potencia aparente de un fármaco cuando se relaciona
con la concentración plasmática total del fármaco. Un aumento de
la fracción libre sube la presión de propulsión al lugar del efecto del
fármaco. Por tanto, un cambio en la fracción libre aumenta la con-
centración en el lugar del efecto. Debido a este cambio aparente de
potencia, un descenso de la unión a proteínas puede disminuir la
dosis necesaria para producir determinado efecto de un fármaco,
incluso en ausencia de otros cambios farmacocinéticos.
Estereoquímica
Los análisis farmacocinéticos habituales describen fármacos ficticios.
Por ejemplo, la farmacocinética y farmacodinámica de tiopental, fen-
tanilo y midazolan describen fármacos que no existen. La razón es
que la mayoría de los anestésicos son quirales (enantiómeros) y se
suministran comomezclas racémicas.El cuerpo es un ambiente quiral
y los fármacos interaccionan de modo esteroespecífico con enzimas,
proteínas y receptores. La farmacocinética y farmacodinámica de los
enantiómeros puede ser diferente. Se han estudiado con más detalle
Principios básicos de farmacología
253
9
Sección II
Farmacología y anestesia
© ELSEVIER. Fotocopiar sin autorización es un delito
Figura 9-10
Relación entre concentración plasmática de proteína como
fracción de la concentración asociada a una unión del 50% y fracción del
fármaco no unida a proteína. Se trata de otro ejemplo de la ecuación de
saturación de la
figura 9-5 .Figura 9-11
Relación entre cambios en la concentración de proteína y
cambios en la fracción libre que depende de la fracción libre en plasma normal
con dosis clínicas habituales. Para un fármaco que no está unido (fracción
libre=1) no se produce cambio en la fracción libre tras un cambio en la
concentración de proteína. En el caso de un fármaco muy unido con una
fracción libre cercana a 0, un cambio en la proteína produce un cambio casi
inversamente proporcional en la fracción libre. (
Modificado de Shafer S:
Principles of pharmacokinetics and pharmacodinamics
. En
Longnecker DE,
Tinker JH, Morgan GE [eds.]:
Principles and Practice of Anesthesiology,
2.
a
ed.
St. Louis, Mosby-Year Book, 1997.
)