








 91 / 2894
91 / 2894

anestésic
o 250como con la especie. Sin embargo, en personas aneste-
siadas con halotano y N
2
O, el umbral de FSC para los cambios EEG
iniciale
s 252es similar al observado en investigaciones animales.
Modelos de isquemia cerebral
Se ha avanzado mucho en el conocimiento de las diferencias entre
la isquemia cerebral global, que se produce durante una parada
cardíaca, y la isquemia cerebral incompleta, que puede ocurrir
durante la oclusión de un vaso sanguíneo principal o la hipoten-
sión grave. Sin embargo, desde la perspectiva del clínico, la diferen-
cia importante es que el flujo sanguíneo residual durante la isquemia
incompleta puede producir un suministro de oxígeno suficiente
como para permitir alguna generación de ATP y, por ende, inte-
rrumpir el fallo de membrana catastrófico e irreversible que se
produce en cuestión de minutos durante la isquemia cerebral com-
pleta normotérmica. Esta diferencia en la tasa de fracaso del aporte
energétic
o 253,254 ( fig. 3-15) puede dar lugar a una tolerancia mani-
fiesta para la isquemia cerebral focal o incompleta mayor que para
la isquemia global completa (p. ej., una parada cardíaca).
Fallo energético y excitotoxicidad
El fallo energético es el acontecimiento central que se produce
durante la isquemia cerebra
l 255 .Se requiere ATP para el manteni-
miento de un gradiente iónico de membrana normal, y al fallo
energético le sigue rápidamente una despolarización de la mem-
brana y entrada de sodio y calcio dentro de la neurona. A conti-
nuación, se activan canales de calcio dependientes de voltaje y el
calcio consigue entrar en el citosol. La despolarización de las ter-
minales presinápticas también provoca la liberación de cantidades
masivas de neurotransmisores excitadores, en particular de gluta-
mato, en el interior de la hendidura sináptica. La activación de
receptores glutamatérgicos, el
N
-metil-d-aspartato (NMDA) y el
ácido
a
-amino-3-hidroxi-5-metil-4-isoxazopropiónico (AMPA) se
añade al influjo de Na
+
y Ca
2+
( fig. 3-16). El inicio de la señalización
celular mediante la activación de receptores metabotrópicos lleva
a la liberación del Ca
2+
almacenado desde el retículo endoplásmico
a través de los receptores del inositol 1,4,5-trifosfato (IP
3
).Al influjo
iónico le acompaña la entrada de agua, por lo que se produce
rápidamente un edema neuronal después de la despolarización de
la membrana. La lesión que se inicia por la actividad excesiva del
receptor de glutamato se denomina excitotoxicidad.
El calcio es un segundo mensajero ubicuo en las células y
constituye un cofactor necesario para la activación de una serie de
sistemas enzimáticos. El incremento rápido e incontrolado de los
niveles citosólicos de Ca
2+
activa varios procesos celulares que
Fisiología cerebral y efectos de los anestésicos
91
3
Sección I
Fisiología y anestesia
© ELSEVIER. Fotocopiar sin autorización es un delito
Figura 3-15
Comparación de las tasas de fallo en el aporte energético
(adenosín trifosfato) en la isquemia global completa (producida por
decapitación en perro
s 254) y en la isquemia focal incompleta (oclusión de la
arteria cerebral media [ACM] en mono
s 253 ). En presencia de FSC residual, el
fallo en el suministro de energía se demora sustancialmente.
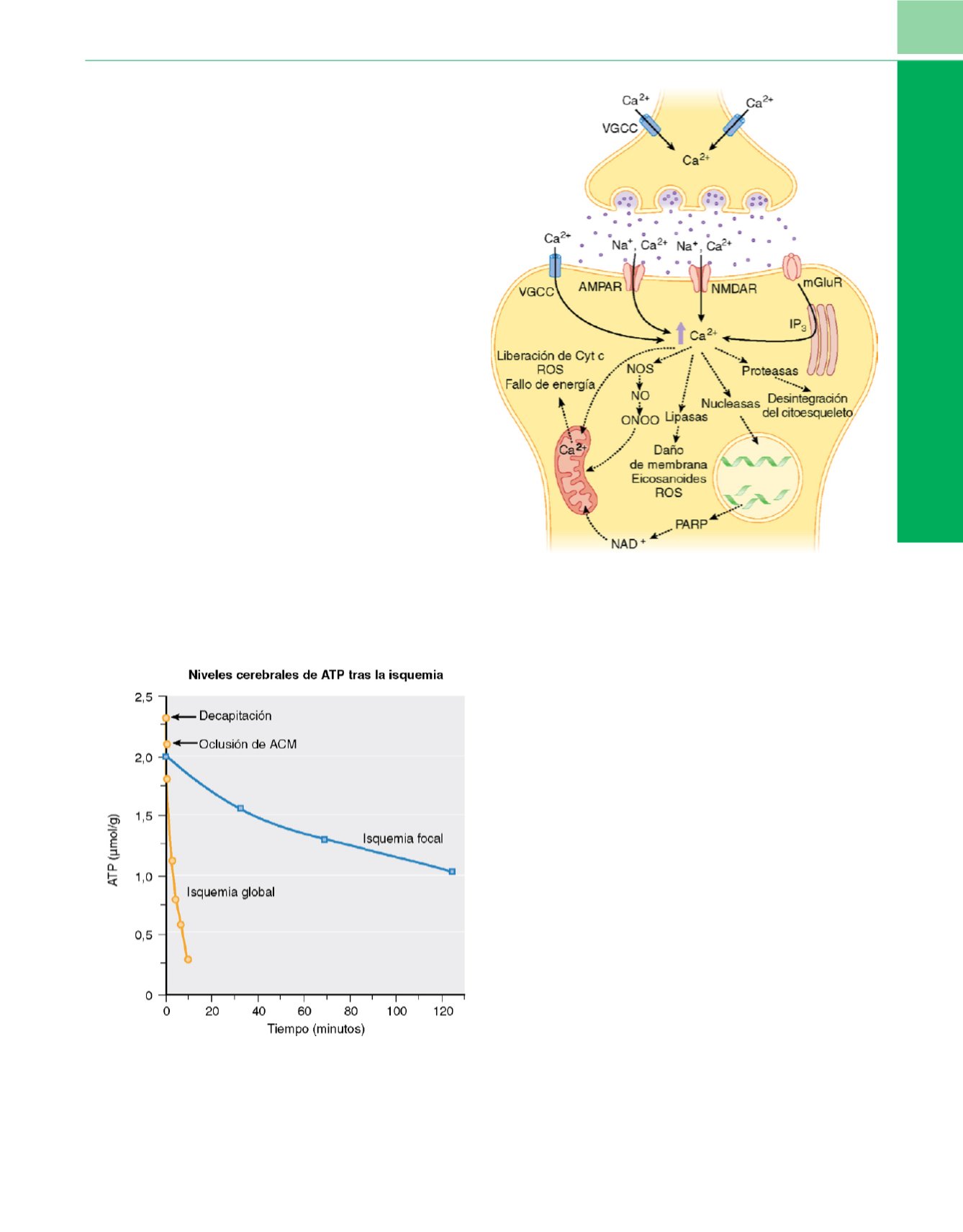
Figura 3-16
Durante la isquemia, la depleción de adenosín trifosfato conlleva
una despolarización neuronal y la consiguiente liberación de cantidades
anormalmente elevadas de neurotransmisores, en especial de glutamato. La
estimulación excesiva de canales dependientes de ligandos y la apertura
simultánea de canales de Ca
2+
dependientes del voltaje permite la entrada
rápida de Ca
2+
al interior de las neuronas. La estimulación de receptores
metabotrópicos de glutamato genera inositol 1, 4, 5-trifosfato (IP
3
), que provoca
la liberación de Ca
2+
desde el retículo endoplasmático (RE)/mitocondrias. La
activación del subconjunto de receptores de glutamato ligados al ácido
a
-amino-3-hidroxi-5-metil-4-isoxazopropiónico (AMPAR) también permite la
entrada excesiva de sodio (Na
+
). El exceso de Ca
2+
libre produce la activación de
numerosas enzimas: la activación de proteasas causa la rotura del citoesqueleto
de la neurona; las lipasas dañan los lípidos de la membrana plasmática y liberan
ácido araquidónico, que es metabolizado por la ciclooxigenasa y por
lipooxigenasas para producir radicales libres y otros mediadores del daño
celular; la activación de la sintetasa de óxido nítrico (NOS) provoca la liberación
de óxido nítrico (NO) y, como consecuencia, la generación de peroxinitrito
(ONOO), un radical libre altamente reactivo; y las endonucleasas activadas dañan
al ADN, por lo que hacen que las neuronas sean susceptibles a la apoptosis. El
daño mitocondrial conlleva un fallo energético, la generación de radicales libres
y la liberación de citocromo
c
(Cyt c) desde la mitocondria; lo último es uno de
los medios por los cuales se inicia la apoptosis neuronal. mGluR, receptor
metabotrópico de glutamato; NMDAR, receptor de
N
-metil-
d
-aspartato; PARP,
poli-ADP-ribosa polimerasa; ROS, especies reactivas de oxígeno; VGCC, canales
de calcio dependientes del voltaje.