








 92 / 2894
92 / 2894

contribuyen al daño. Las proteínas citoesqueléticas, como la actina,
son degradadas por las proteasas activadas. Estas enzimas también
degradan una cantidad de constituyentes proteicos de la mem-
brana. Las lipasas atacan a los lípidos celulares y producen daño de
membrana. Una lipasa importante, la fosfolipasa A
2
, libera ácidos
grasos de las membranas, como el ácido araquidónico. El metabo-
lismo del ácido araquidónico en prostaglandinas y leucotrienos por
la ciclooxigenasa y la lipooxigenasa se acompaña de la generación
de radicales libres superóxido. Éste, combinado con otros radicales
libres generados como respuesta al daño mitocondrial, puede llevar
a la peroxidación lipídica y al daño de membrana. Las prosta-
glandinas y los leucotrienos evocan también una respuesta infla-
matoria y son potentes agentes quimiotácticos. La activación
plaquetaria dentro de la microvascularización cerebral, así como la
entrada de leucocitos a las áreas dañadas, agravan el daño isqué-
mico por oclusión de la vascularización.
La lesión del ADN es también un evento importante en el
daño neuronal isquémico. La generación de radicales libres desde el
metabolismo del ácido araquidónico, desde las mitocondrias lesio-
nadas y desde la producción de peroxinitrito a partir del NOconduce
a una lesión oxidativa del ADN. La activación de endonucleasas
produce también la ruptura de las cadenas de ADN. En circuns-
tancias normales, el daño sobre el ADN provoca la activación de la
poli-ADP-ribosa-polimerasa (PARP), una enzima que participa en
la reparación del ADN. Si hay un daño excesivo del ADN, la activi-
dad de la PARP se incrementa drásticamente y esta actividad
aumentada puede conducir a la depleción de nicotinamida-adenín-
dinucleótido (NAD), un sustrato de la PARP. La NAD también es
una coenzima importante en el metabolismo energético y su deple-
ción exacerba aún más el fallo energético.
La formación de lactato es un elemento adicional del proceso
fisiopatológico. El ácido láctico se forma como resultado de la
glucólisis anaerobia que se lleva a cabo después del fallo en el
suministro de oxígeno. La disminución asociada del pH contribuye
al deterioro del medioambiente intracelular. Un aumento de los
niveles séricos de glucosa previos a la isquemia puede acelerar este
proceso al proveer sustrato adicional para la glucólisis anaerobia.
El NO, que ha surgido como un probable mediador de los
cambios en el FSC en muchas situaciones fisiológicas normales
(v. las secciones anteriores), tiene también relevancia en la fisiopa-
tología de la isquemia. De hecho, el NO es un radical libre débil
que puede llevar a la generación de especies más reactivas (pero-
xinitrito) y es una «sustancia asesina» empleada por los macrófa-
gos. En la isquemia cerebral, el NO es probablemente tanto amigo
como enemigo. Es posible que durante un período de isquemia
focal, el efecto vasodilatador del NO (probablemente NO elabo-
rado en el endotelio) sirva para aumentar el FSC colateral. Sin
embargo, en la fase postisquémica el NO (probablemente NO
inducido de origen neuronal) contribuye al daño neuronal.
De forma colectiva, la activación simultánea y no regulada
de una serie de vías celulares sobrepasa los procesos reparativos y
restauradores en el interior de la neurona, y finalmente conduce a
la muerte neuronal.
Naturaleza de la muerte neuronal
La muerte neuronal que se produce como respuesta a estos proce-
sos ha sido clasificada como de naturaleza necrótica o apoptótica
(v. cap. 88). La muerte neuronal necrótica, mediada por excitotoxi-
cidad, se caracteriza por un edema celular rápido, condensación y
picnosis del núcleo, así como edema de las mitocondrias y del
retículo endoplásmico. Una peculiaridad de estas neuronas necró-
ticas es la presencia de un citoplasma acidófil
o 256 .La muerte neu-
ronal necrótica provoca una infiltración cerebral local de células
inflamatorias. Una consecuencia de esta inflamación es una canti-
dad considerable de daño colateral.
En una serie de modelos de isquemia cerebral también se ha
observado la presencia de apoptosis neuronal, una forma de suici-
dio celular. La apoptosis se caracteriza por condensación de la
cromatina, involución de la membrana celular, inflamación de las
mitocondrias y desestructuración celular. En las últimas fases del
proceso apoptótico, las neuronas se fragmentan en varios cuerpos
apoptóticos que son luego eliminados del parénquima cerebra
l 256 .La ausencia de una respuesta inflamatoria sustancial a la muerte
apoptótica limita la lesión a las neuronas adyacentes que han sobre-
vivido a la lesión isquémica inicial.
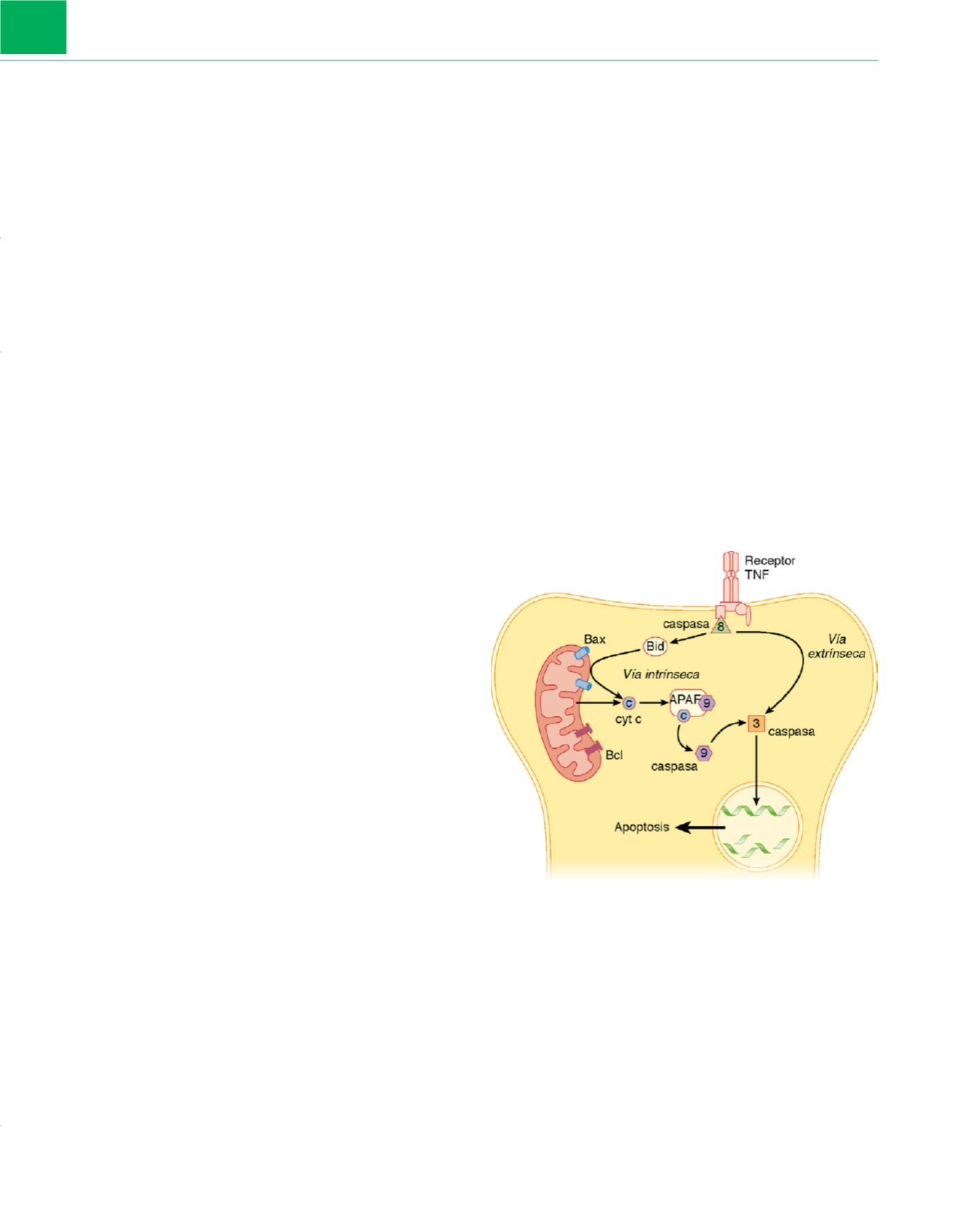
Se ha descrito una serie de vías bioquímicas que conducen a
la apoptosis. La apoptosis iniciada por la liberación de citocromo
c
desde las mitocondrias dañadas ha sido la más estudiada
( fig. 3-17).
El citocromo
c
está restringido del citoplasma por la membrana
mitocondrial extern
a 257. Cuando se dañan las mitocondrias, los
poros contenidos en la membrana mitocondrial externa permiten
la liberación de citocromo
c
en el citoplasma, donde interactúa con
la procaspasa-9 y el factor activador de la apoptosis (APAF) para
producir un apoptosoma. La procaspasa-9 sufre una activación
mediante escisión proteolítica. La caspasa-9 activada activa poste-
riormente la caspasa-3. Esta última sirve como ejecutor de la apop-
tosis al fragmentar un número de sustratos proteicos que son esen
ciales en la reparación del ADN (como la PARP). La activación de
la caspasa-3 también puede producirse mediante señalización infla-
matoria a través de la activación del factor de necrosis tumoral-
a
(TNF-
a
) y caspasa-
8 258. Hay que señalar que el daño neuronal pro-
ducido como respuesta a la isquemia no puede ser fácilmente cata-
logado como isquémico o apotótico. La naturaleza de la muerte
92
Fisiología y anestesia
I
Figura 3-17
Procesos celulares que conducen a la apoptosis neuronal. El
citocromo
c
(cyt c), normalmente restringido al espacio entre las membranas
mitocondrial externa e interna, se libera como respuesta al daño mitocondrial.
El citocromo
c,
combinado con el factor activador de la apoptosis (APAF), activa
la caspasa-9 mediante escisión enzimática. La caspasa-9 activada conduce a la
activación de la caspasa-3. Esta enzima escinde varios sustratos, incluidos los
necesarios para reparar el ADN. Dentro de la mitocondria el Bax aumenta y el
Bcl previene la liberación de citocromo
c
. También puede producirse la
liberación de citocromo
c
por Bid, una sustancia que es activada por la
caspasa-8 a través de la señalización por el factor de necrosis tumoral (TNF).
Además, la caspasa-8 puede activar directamente a la caspasa-3. La activación
excesiva de poli-ADP-ribosa polimerasa (PARP), una enzima imprescindible en la
reparación del ADN, depleciona los depósitos celulares de nicotinamida
adenina dinucleótido oxidado (NAD
+
). La depleción de NAD
+
exacerba aún más
el fallo energético porque éste es básico en el metabolismo energético.