








 1570 / 2894
1570 / 2894

secundarios intolerable
s 64 .La adaptación inducida por opioides
sucede a múltiples niveles en el sistema nervioso central y otros
órganos; comienza con la modulación directa de la señalización del
receptor opioide y se extiende a una compleja red neuronal, inclu-
yendo las conductas aprendidas. Los mecanismos propuestos impli-
cados en la tolerancia farmacodinámica son el desacoplamiento del
receptor opioide y la proteína G, la disminución de receptores por
internalización/reciclaje y el aumento de la sensibilidad del receptor
NDMA (v.
fig. 48-3, panel inferior
) 61,65. Además, la farmacocinética
(p. ej., alteración en la distribución o metabolismo de los opioides)
y la tolerancia aprendida (p. ej., habilidades compensatorias desarro-
lladas durante una intoxicación leve) y un aumento de la estimula-
ción nociceptiva por factores de crecimiento tumoral, inflamación o
formación de neuromas son posibles razones para incrementar las
necesidades de dosi
s 62,66 .Existe una falta de cuidadosos estudios
clínicos controlados que demuestren de forma inequívoca la toleran-
cia farmacodinámica a la analgesia por opioides en paciente
s 67 .Actualmente se está debatiendo si los opioides pueden para-
dójicamente inducir hiperalgesi
a 68 .Sin embargo, en un escrutinio
más detallado de los datos disponibles parece que la mayoría de los
estudios han demostrado en realidad hiperalgesia inducida por
retirada, un fenómeno bien conocido después del cese abrupto de
opioides (v. también más adelante la sección «Manejo perioperato-
rio de pacientes con dolor crónico»). A dosis ultraelevadas, ocasio-
nalmente utilizadas en pacientes con dolor oncológico extremo, se
han observado casos singulares de alodinia, que se han atribuido a
los efectos neuroexcitatorios de los metabolitos de los opioide
s 69 .No existe una prueba concluyente de que aparezca hiperalgesia
durante la administración crónica o perioperatoria de dosis habi-
tuales de opioides en paciente
s 68 .Los opioides son efectivos en la periferia (p. ej., por administra-
ción tópica o intraarticular, particularmente en tejidos inflamados), en
el neuroeje (por administración intratecal, epidural o intracerebroven-
tricular) y sistémicamente (administración intravenosa, oral, subcutá-
nea, sublingual o transdérmica
) 61,70 .La elección clínica de un compuesto
en particular o su formulación se basa en consideraciones farmacociné-
ticas (vía de administración, inicio deseado o duración, lipofilidad) y en
los efectos secundarios asociados con la respectiva vía de administración
del fármac
o 71,72 .Las dosis dependen de las características del paciente,
del tipo de dolor y de la vía de administración. Los opioides adminis-
trados por vía sistémica y espinal pueden producir efectos secundarios
similares, dependiendo de la posología y la redistribución sistémica/
rostral. Para la aplicación intratecal se prefieren fármacos lipofílicos, ya
que son atrapados en lamédula espinal y esmenos probable quemigren
hasta el cerebro a través del líquido cefalorraquídeo. Los efectos secun-
darios adversos pueden minimizarse mediante una cuidadosa dosifica-
ción y una monitorización intensa del paciente, o pueden tratarse con
otros medicamentos (antieméticos, laxantes) o con naloxona. No se han
publicado efectos adversos significativos con la aplicación periférica (p.
ej., tópica) de pequeñas dosis de opioides inactivas sistémicamente. La
investigación actual tiene comometa el desarrollo de opioides aplicables
de forma sistémica con un acceso restringido al cerebr
o 11,73 .Los opioides son los fármacos más eficaces para tratar el dolor
agudo intenso y el dolor oncológico crónico. El uso de opioides en
el dolor crónico no oncológico (p. ej., neuropático, musculoesquelé-
tico) no está apoyado de forma inequívoca por ensayos clínicos
aleatorizados (ECA) ciegos y con una potencia adecuada, y los datos
epidemiológicos sugieren que los opioides no mejoran la calidad de
vida o la capacidad funciona
l 74. Hasta ahora, los estudios doble ciego
controlados con placebo han investigado hasta un máximo de dura-
ción del tratamiento de 8 semana
s 75,76. Sólo unos pocos ECA han
requerido una estricta retirada de analgésicos no opioides antes del
estudio para comprobar los efectos de una única medicación con
opioide
s 75,77-80, y algunos otros han examinado una mezcla no espe-
cífica de fármacos opioides y no opioides. Los máximos valores de
reducción del dolor fueron del 17% en comparación con placebo, un
efecto clínicamente no significativo. Parámetros de resultados más
importantes como los factores psicosociales, la calidad de vida y el
estado funcional han sido investigados enmuy pocos ensayo
s 75-79,81,82 .Sólo uno de los estudios que utilizó solamente opioide
s 78y dos que
utilizaron fármacos mixto
s 82,83demostraron una discreta mejoría.
Los ya conocidos efectos secundarios adversos de los opioides
(náuseas, sedación, estreñimiento, mareo, etc.) se observaron en
todos estos estudios y provocaron el abandono de un número signi-
ficativo (hasta el 58%) de sujetos. Por esto, consistente con la
1570
Anestesia por subespecialidades en el adulto
IV
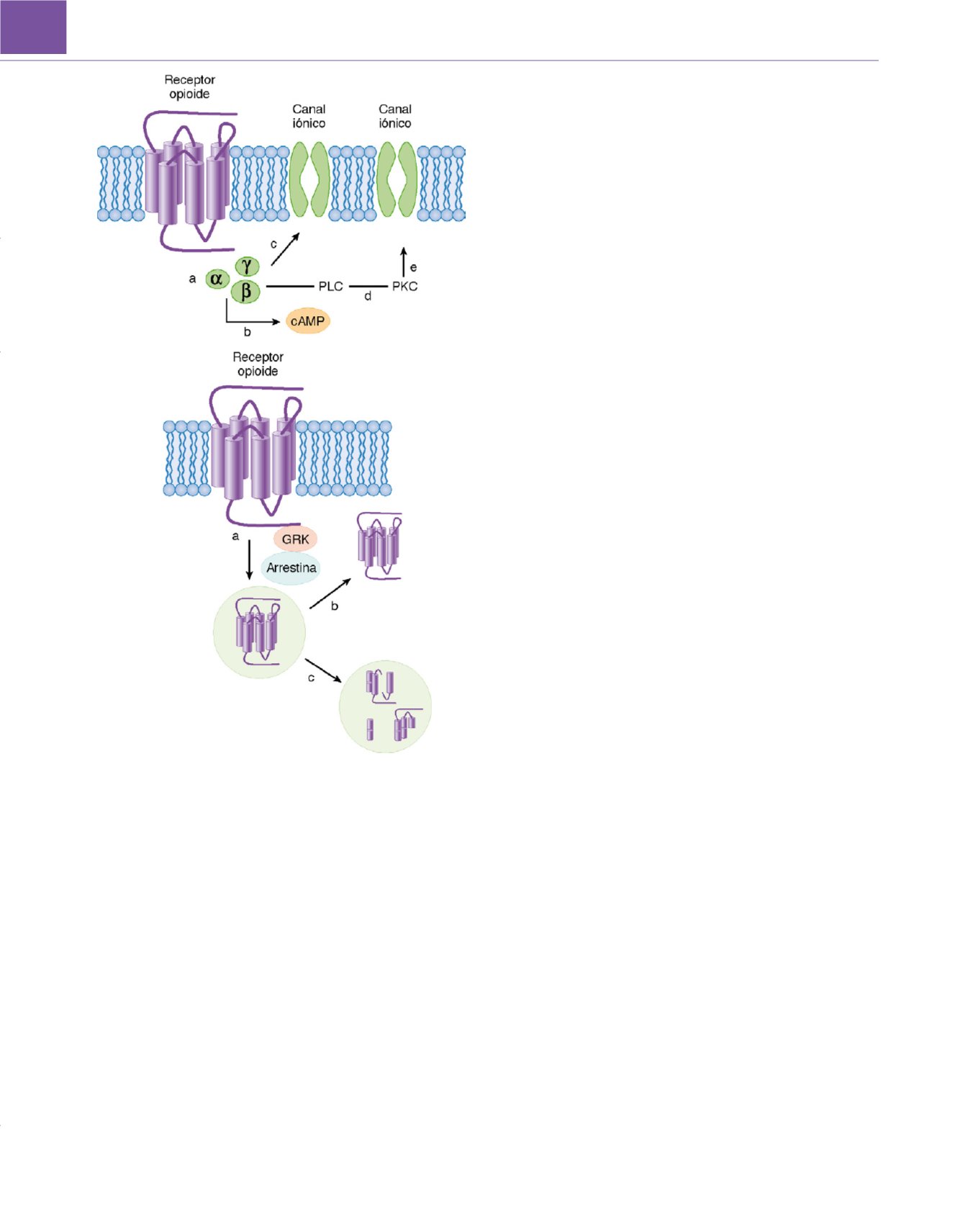
Figura 48-3
Señalización y reciclaje del receptor opioide.
Panel superior,
los ligandos opioides inducen un cambio conformacional en el receptor que
permite el acoplamiento de proteínas G al receptor. La proteína G
heterotrimérica se disocia en las subunidades activas G
a
y G
bg
(a) que
pueden inhibir las adenilciclasas y disminuir el adenosín monofosfato cíclico
(AMPc) (b), reduciendo la conductancia de los canales de Ca
2+
dependientes
del voltaje o abriendo los canales de K
+
de rectificación (c). Además, las vías
de la fosfolipasa C (PLC) y la fosfocinasa C (PKC) pueden activarse (d) para
modular la actividad del canal de Ca
2+
en la membrana plasmática (e).
Panel
inferior,
la desensibilización del receptor opioide y su movimiento son
activados por los receptores de cinasas acoplados a proteínas G (GRK).
Después de la unión de la arrestina, el receptor se encuentra en un estado
desensibilizado en la membrana plasmática (a). Los receptores de unión de la
arrestina pueden entonces ser internalizados a través de una vía dependiente
de clatrina y ser o bien reciclados hasta la superficie celular (b) o bien
degradados en los lisosomas (c). (
Adaptada de Zöllner C, Stein C: Opioids.
Handb Exp Pharmacol
177:31-63, 2007
.)