








 404 / 2894
404 / 2894

Diferencias de desarrollo en lactantes y niños
Las enzimas de fase 1 y 2 empiezan a madurar en el período pos-
natal (v. también cap. 72), pero las concentraciones pueden ser
insuficientes en algún caso para que se produzca toxicidad si no se
controla con cuidado la administración de fármacos. Por ejemplo,
la glucuronización insuficiente de la bilirrubina en el recién nacido
puede producir hiperbilirrubinemia y el síndrome del «niño gris»
por aumento de la concentración de cloramfenico
l 9,10 .Con el cre-
cimiento, parece que es mayor la afectación de enzimas de fase 1
que de fase 2. La isoforma más expresada en el hígado fetal es
CYP3A7, que alcanza el máximo después del nacimiento y
disminuye rápidamente hasta concentraciones indetectables en la
mayoría de los adulto
s 11 .A las pocas horas del nacimiento, aumenta
la actividad de CYP2E1, y después las de CYP2D6, CYP3A4,
CYP2C9 y CYP2C19. CYP1A2 es la última isoforma que aparece,
alrededor de los 2 meses de edad. CYP2C9 y CYP2C19 son las
responsables de la biotransformación de la difenilhidantoína. La
semivida de ésta se prolonga hasta 75 horas en recién nacidos, pero
disminuye a 20 horas en recién nacidos a término durante la
primera semana de vida y a 8 horas en la segunda seman
a 12 .Farmacogenética del metabolismo
de los fármacos
La evolución de la genética ha tenido un impacto importante en el
metabolismo de los fármacos y ha proporcionado las herramientas
experimentales para demostrar la base genética de la variabilidad
entre individuos en la farmacocinética, metabolismo y toxicidad
de los fármacos en el ser human
o 13 .Aunque con frecuencia se ha
creído que los CYP son los responsables de desactivar los compues-
tos tóxicos, también se encargan de la activación metabólica de
fármacos y sustancias químicas a formas tóxicas. Cualquier factor
que influye en el metabolismo puede afectar a la toxicidad. Como
ya se ha comentado, hay muchos factores que pueden afectar a la
biotransformación, como la vía y la frecuencia de administración,
la exposición a otras sustancias químicas, el sexo, la edad, la dieta
y la genética. El concepto de farmacogenética deriva de la obser-
vación clínica de que algunos pacientes tenían concentraciones de
fármacos muy bajas o muy altas en sangre y orina y que esta carac-
terística era hereditaria. Muy pronto se identificaron las enzimas
que metabolizan los fármacos y los genes que codifican las proteí-
nas y las secuencias de ADN dentro de los genes. La mayor parte
de las características farmacogenéticas iniciales eran monogénicas,
afectando a un solo gen, y la mayoría estaban producidas por
polimorfismo genético.
El descubrimiento de que la alteración de la hidrólisis de la
succinilcolina en fase 1 debida a butirilcolinesterasa era heredita-
ria, supuso un estímulo para el desarrollo de la farmacogenética.
Cerca de 1 de cada 3.500 personas de raza blanca es homocigota
para el gen que codifica una forma atípica de butirilcolinesterasa.
Estos afectados tienen menor capacidad para hidrolizar la succini-
lcolina, y por tanto los efectos del bloqueo neuromuscular se man-
tienen más tiempo en ello
s 14,15. En estudios más recientes se ha
demostrado que la CYP2D6 representa uno de los ejemplos de
variación farmacogenética en el metabolismo de los fármacos
mejor comprendido. La codeína, el metroprolol, la nortriptilina, el
dextrometorfán, la debrisoquina y la esparteína son sustratos de
CYP2D
6 16 .Se descubrió que del 5% al 10% de los pacientes blancos
no metabolizaban adecuadamente el antihipertensivo debriso-
quin
a 16y el antiarrítmico esparteín
a 17, y que mantenían concentra-
ciones del fármaco original altas en plasma y concentración
urinaria baja de metabolitos. Esta característica se hereda de forma
autosómica recesiv
a 17,18. Se ha clonado el ADNc del gen que codi-
fica CYP2D6 y se han identificado diversas variantes genéticas
responsables de la actividad deficiente de esta enzima. Otros
pacientes tienen copias múltiples de las formas activas de CYP2D6
que producen una eliminación rápida del fármaco y por tanto
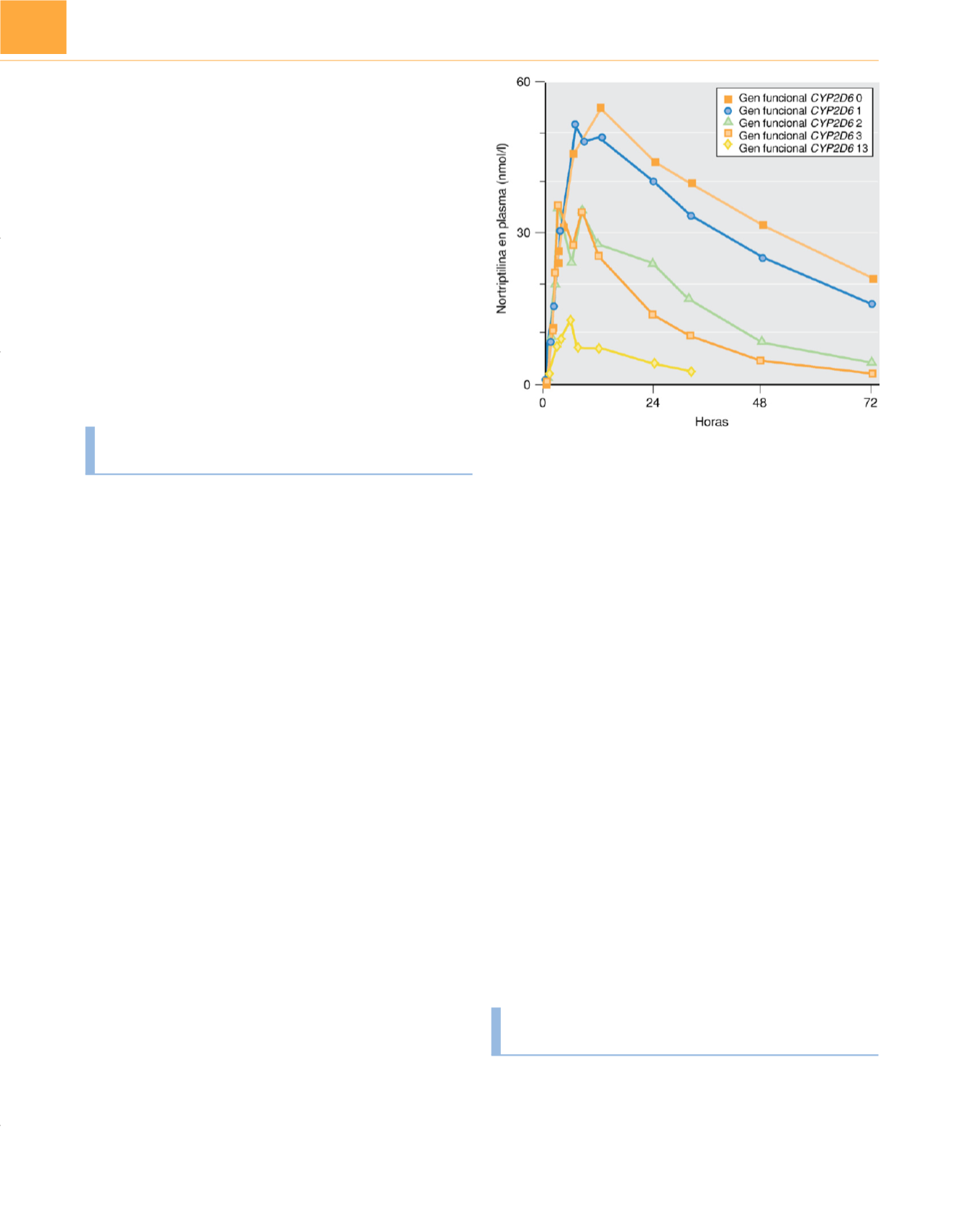
concentraciones subterapéuticas. Es el caso del antidepresivo nor-
triptilina (
fig. 14-5 ). Se han descrito muchos otros ejemplos de
metabolizadores deficientes de fármacos por variantes genéticas
de otras isoformas de CYP. Existen variantes de CYP2C9 que meta-
bolizan mal la warfarina y la difenilhidantoína, llevando a concen-
traciones tóxicas de estos fármacos. También los polimorfismos de
la enzima de fase 2 NAT pueden producir diferencias bimodales
en la
N-
acetilación e inactivación del antituberculoso isoniazida
( fig. 14-6). En estudios de clonación molecular más recientes se ha
demostrado que en los seres humanos hay dos genes,
NAT1
y
NAT2,
y que el polimorfismo genotípico frecuente responsable de
la variación farmacogenética en el metabolismo de la isoniazida se
debe al gen
NAT
2 19. La frecuencia de cada fenotipo de acetilación
depende de la raza, pero no del sexo ni de la edad. En inuits y
japoneses se observa acetilación rápida, mientras que la acetilación
lenta predomina en escandinavos y norteafricanos blanco
s 20 .Otro
ejemplo del papel de la genética en el metabolismo de fase 2 es el
antineoplásico azatioprina, un precursor que se convierte en
6-mercaptopurina activa. Las tiopurinas como la 6-mercaptopu-
rina están metabolizadas por la tiopurina-
S
-metiltransferasa
(TPMT
) 21 .Esta actividad enzimática se hereda de forma autosó-
mica codominante. Los homocigotos para los alelos que codifican
la TPMT inactiva y tratados con dosis estándar de azatioprina
tienen riesgo de desarrollar pancitopenia grave.
Farmacogenómica del metabolismo
de los fármacos
La
farmacogenómica
es el resultado de la convergencia de la far-
macogenética y la genómica humana. Este término se utiliza para
describir la influencia de las variaciones de secuencias de ADN
404
Farmacología y anestesia
II
Figura 14-5
Farmacogenética de la nortriptilina. Concentraciones plasmáticas
medias de nortriptilina después de una dosis oral única de 25mg en individuos
con genes funcionales CYP2D6 0, 1, 2, 3 y 13.
(Adaptada de Weinshilboum R:
Inheritance and drug response.
N Engl J Med
348:529-537, 2003.)