








 596 / 2894
596 / 2894

Por tanto, se puede calcular la velocidad de infusión necesaria,
I(t), para una determinada concentración deseada, C
T
(t), y una deter-
minada farmacocinética, D(t), utilizando las mismas herramientas
que se usan para calcular la farmacocinética inicial. Por desgracia, esta
solución puede requerir velocidades de infusión negativas, lo que
obviamente es imposible.Dado que no podemos succionar el fármaco
del cuerpo (es decir, administrar infusiones negativas), tenemos que
limitarnos a concentraciones plasmáticas a lo largo del tiempo que se
puedan alcanzar con velocidades de infusión no negativas.
El modelo farmacocinético estándar tiene un gran defecto,
pues asume que tras la administración de un bolo, éste se mezcla por
completo en el compartimento central, de modo que la concentra-
ción pico se alcanza precisamente en el momento 0. En realidad, el
fármaco tarda 30-45 segundos en ir desde el sitio de la administra-
ción venosa hasta la circulación arterial. Este defecto puede parecer
insignificante, pero puede ocasionar problemas cuando se trata de
relacionar el efecto del fármaco después de la administración de un
bolo con la concentración del fármaco en el cuerp
o 10 .Los investiga-
dores estánmodificando los modelos farmacocinéticos poliexponen-
ciales para ofrecer otros más exactos de la concentración plasmática
del fármaco en el primer minuto tras la inyección del bol
o 11 .Consideraciones
farmacodinámicas
La biofase
Aunque la concentración plasmática después de la administración
intravenosa de un fármaco alcanza su máximo casi de forma ins-
tantánea, ningún anestesiólogo se atrevería a intubar a un paciente
justo tras administrarle el bolo de un hipnótico. La razón, por
supuesto, es que, aunque la concentración máxima plasmática se
alcanza casi de inmediato, es necesario más tiempo para que
aumente la concentración del fármaco en el cerebro y produzca la
pérdida de consciencia. Este retraso entre la concentración plas-
mática máxima y la concentración máxima en el cerebro se deno-
mina
histéresis
. Ésta es la traducción clínica de que el plasma no es
el lugar donde el fármaco ejerce su efecto, sino un medio de trans-
porte. Los medicamentos ejercen su efecto farmacológico en la
biofase
, que también se denomina «sitio de efecto». La biofase es
el medio en el que el fármaco ejerce directamente su efecto en el
cuerpo, e incluye las membranas, los receptores y las enzimas.
Hay dos razones por las que no se puede medir la concen-
tración del fármaco en la biofase. Primera, porque normalmente es
inaccesible, al menos en los seres humanos. Segunda, porque
aunque se pudieran tomar muestras de tejido, la concentración del
fármaco en el ambiente microscópico de las moléculas receptoras
no es la misma que la concentración medida, por ejemplo, en el
cerebro homogeneizado. Aunque en realidad no es posible medir
de forma exacta la concentración del fármaco en la biofase, se
puede caracterizar la evolución en el tiempo del efecto del fármaco
con medidas rápidas de la acción del mismo. Si se conoce la evolu-
ción en el tiempo del efecto del fármaco, podemos utilizar modelos
matemáticos para saber la velocidad del flujo de salida y de entrada
del fármaco en la biofase (o en el «sitio de efecto»
) 12,13y su concen-
tración aparente en la biofase, usando la concentración plasmática
en el equilibrio estacionario que produciría ese mismo efecto.
Las medidas del efecto que se utilizan para predecir la evo-
lución en el tiempo del fármaco entre el plasma y la biofase variarán
en función del fármaco que se esté evaluando. Para los bloqueantes
neuromusculares, la contracción unitaria es una medida ideal del
efecto. Para los opioides y los hipnóticos, dicha medida resulta más
complicada. El efecto deseado de los opioides es la analgesia, una
medida subjetiva y difícil de monitorizar y cuantificar en cada
momento. De igual forma, hasta hace poco no existía para los
hipnóticos un método válido que evaluase el efecto del fármaco.
Por estos motivos los investigadores han convertido el EEG en la
medida objetiva del efecto de los opioides y los hipnóticos. Aunque
el EEG no mide el efecto deseado de estos fármacos, se asume que
los cambios en el mismo reflejarán al menos la evolución en
el tiempo del efecto clínico. Por tanto, se ha utilizado el EEG
para definir la evolución en el tiempo del equilibrio entre el
plasma y la biofase para los opioides y los hipnóticos. Numerosos
596
Farmacología y anestesia
II
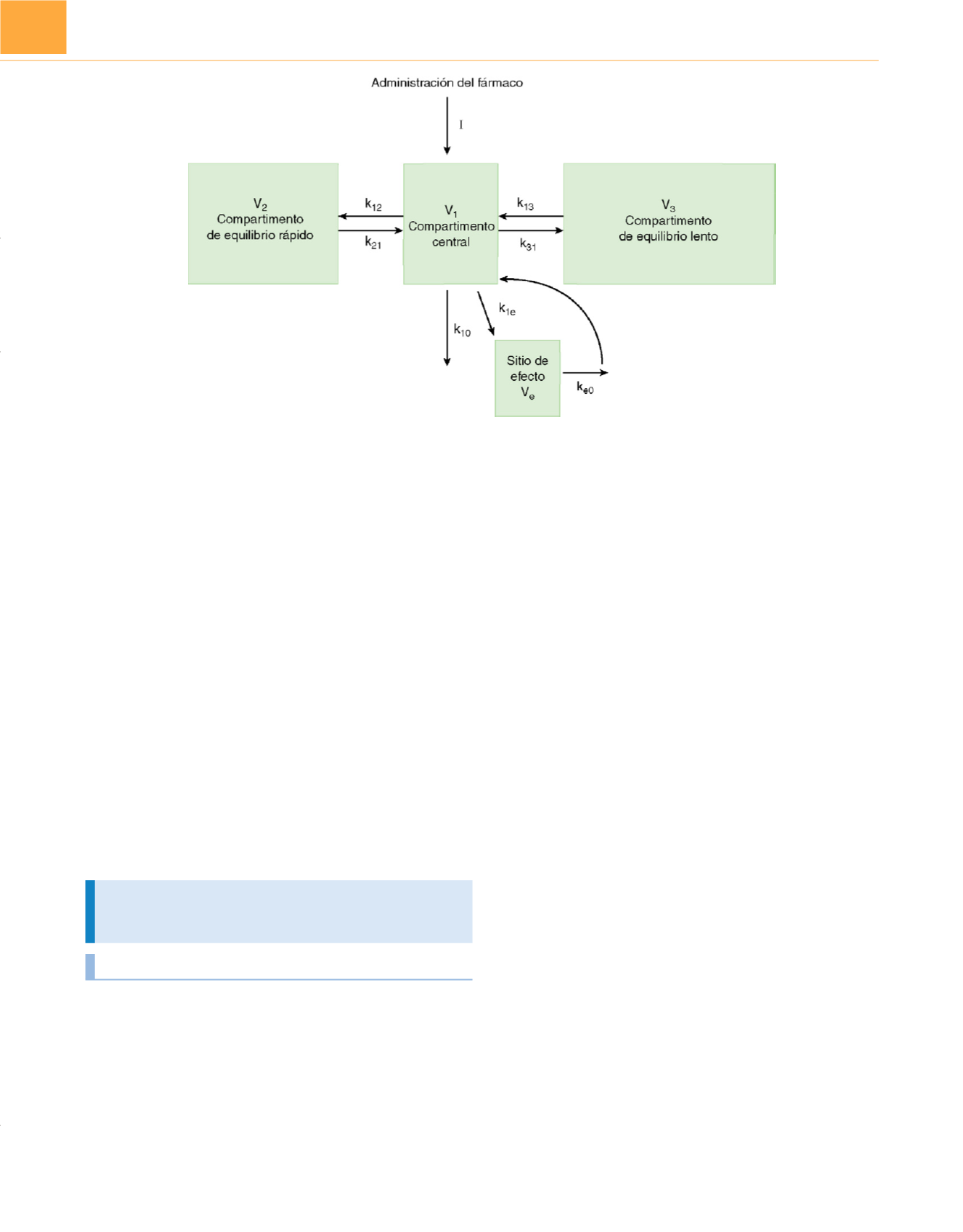
Figura 18-5
Modelo tricompartimental (incluye la biofase) que esquematiza los procesos farmacocinéticos básicos que ocurren tras la administración
intravenosa de un fármaco. I, esquema de dosis en función del tiempo; k
10
, constante de velocidad que refleja todos los procesos que actúan para eliminar el
fármaco de manera irreversible del compartimento central; k
ij
constantes de velocidad intercompartimentales; V
1
, volumen del compartimento central, en
general expresado en litros o en litros por kilogramo.