








 509 / 2894
509 / 2894

relativamente corta de este fármaco; de ahí que se deba tener una
gran precaución al respecto.
Fenciclidinas (ketamina)
Antecedentes históricos
La fenciclidina fue el primer fármaco de esta clase que se usó en
anestesia, pero provocaba efectos secundarios no aceptables. La
ketamina fue sintetizada en 1962 por Stevens, y administrada por
primera vez en seres humanos en 1965 por Corssen y Domino. La
ketamina se comenzó a utilizar en la clínica en 1970, y sigue utili-
zándose en diversos contextos clínicos. Se diferencia de la mayoría
de los otros anestésicos que se usan para la inducción en que tiene
un efecto analgésico significativo. No suele producir depresión car-
diovascular ni respiratori
a 308 ,pero se han encontrado algunos
efectos secundarios psicológicos problemáticos similares a los que
se producen con otras fenciclidinas. La ketamina tiene dos este-
roisómeros: S+ y R−; el isómero S+ es más potente y tiene menos
efectos secundarios. Recientemente ha aumentado el interés por
la ketamina debido al efecto que causa en la hiperalgesia y en la
tolerancia a los opioides, a su uso en el dolor crónico, a sus poten-
ciales efectos neuroprotectores y al aumemto de la popularidad de
la anestesia intravenosa total, y también gracias a la disponibilidad
(en algunos países) de la forma S+ de la ketamina.
Características fisicoquímicas
La ketamina
( fig. 16-14) tiene un peso molecular de 238 kD, es
parcialmente soluble en agua, y forma una sal blanca cristalina con
un p
K
a
de 7,
5 308 .Es 5-10 veces más liposoluble que el tiopenta
l 309 .Metabolismo
Las enzimas microsomales hepáticas metabolizan la ketamina. La
principal vía de metabolización es la
N
-desmetilación para formar
norketamina (metabolito I), que más tarde será hidroxilada para
formar hidroxinorketamina. Estos productos se conjugan a deriva-
dos glucorónidos solubles en agua y se excretan por la orin
a 308 .La
actividad de los principales metabolitos de la ketamina no ha sido
bien estudiada, pero se ha observado que la norketamina tiene una
actividad mucho menor (20-30%) que el compuesto del que se
deriva. Las modelizaciones más recientes de la norketamina sugie-
ren que contribuye en la prolongación de la analgesia producida
tanto por un bolo como por la infusión de ketamin
a 310 .Farmacocinética
Se ha estudiado la farmacocinética de la ketamina tras la adminis-
tración de un bolo a dosis anestésicas (2 a 2,5mg/kg
) 311, después de
dosis subanestésicas (0,25mg/kg
) 311,312y de una infusión continua
(concentración plasmática en el equilibrio estacionario de 2.000ng/
ml). Con independencia de la dosis, la desaparición plasmática de
la ketamina puede describirse como un modelo bicompartimen
ta
l 311. En la
tabla 16-1se muestran los valores farmacocinéticos de
la administración de un bolo. Se ha de señalar que la distribución es
rápida, lo que se traduce en una semivida de distribución relativa-
mente breve, de 11-16 minutos
( fig. 16-15 ). La gran liposolubilidad
de la ketamina se refleja en su volumen de distribución, bastante
grande, de casi 3l/kg. El aclaramiento también es relativamente
rápido (890-1.227ml/min), lo que explica la semivida de eliminación
corta, de 2-3 horas. El aclaramiento corporal total medio (1,4l/min)
es aproximadamente igual al flujo sanguíneo hepático, lo que indica
que las alteraciones en el flujo sanguíneo hepático afectarán al acla-
ramiento. Las dosis bajas de alfentanilo incrementan el volumen de
distribución y el aclaramiento de la ketamina, por lo que aumentarán
su concentración plasmática. Además, el alfentanilo incrementa la
distribución de la ketamina en el cerebr
o 313. El modelo farmacoci-
nético de Clement
s 311ofreció la máxima precisión cuando se utilizó
para administrar dosis bajas de ketamina a voluntarios utilizando un
dispositivo de infusión ajustada a objetivos terapéutico
s 314.
La farmacocinética de los dos isómeros es diferente. La keta-
mina S+ tiene un aclaramiento mayor y un mayor volumen de distri-
bución que la ketamina R
− 315 .En un estudio en el que se analizó la
farmacocinética de la ketamina S+ utilizando un dispositivo de infu-
sión ajustada a objetivos para procedimientos de 1 hora, en combina-
ción con propofol, se observó que se conseguía una mayor precisión
delosparámetrosfarmacocinéticosconunVcmuchomenor(17ml/kg).
También se observó que el aclaramiento de la ketamina no seguía una
distribución normal y que tampoco mostraba una correlación con la
eda
d 316 .El enantiómero S+ parecemás potente en la supresión del EEG
que el enantiómero R− y que la mezcla racémic
a 317.
La ketamina se utiliza cada vez más por vías alternativas,
especialmente por vía oral y mediante un spray intranasal. La
administración por cualquiera de esas dos vías se halla sujeta a un
importante metabolismo de primer paso. La biodisponibilidad tras
Anestésicos intravenosos
509
16
Sección II
Farmacología y anestesia
© ELSEVIER. Fotocopiar sin autorización es un delito
Figura 16-14
Esteroisómeros de la ketamina como viene formulada.
Figura 16-15
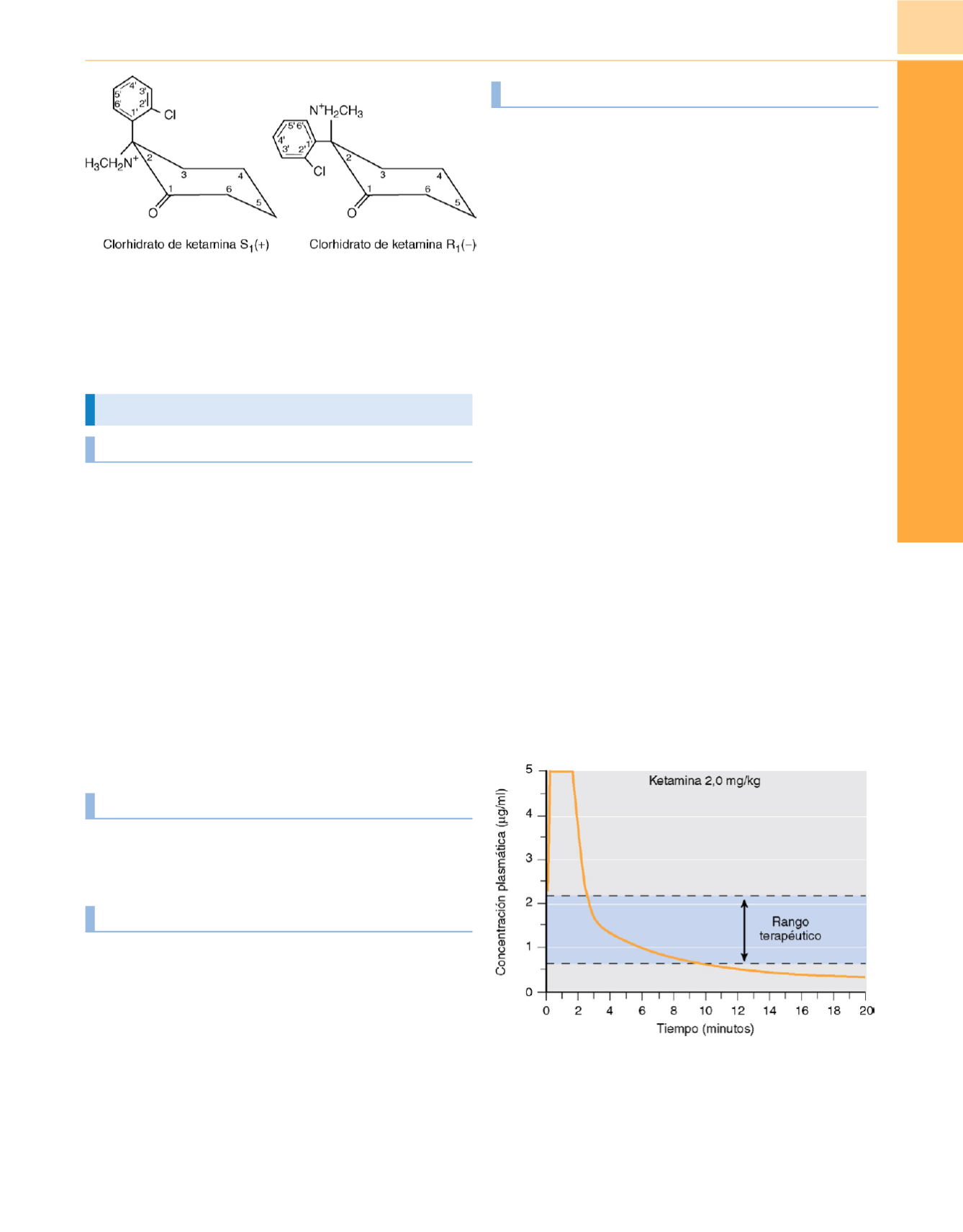
Representación de la evolución en el tiempo de los niveles
plasmáticos de ketamina tras una dosis de inducción de 2mg/kg.
La concentración plasmática necesaria para la hipnosis y la amnesia
durante la cirugía es de 0,7-2,2
m
g/ml; el despertar se suele producir con
niveles por debajo de 0,5
m
g/ml.